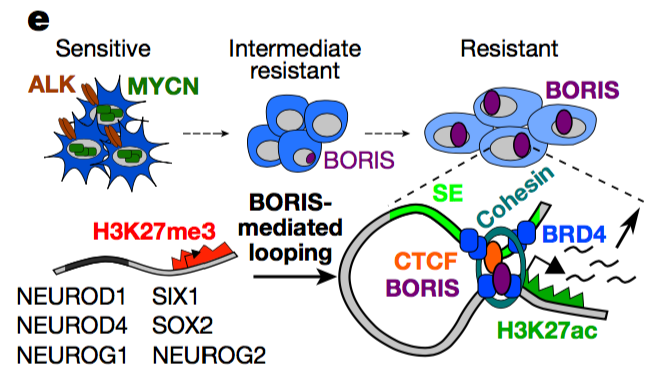
BORIS promotes chromatin regulatory interactions in treatment-resistant cancer cells.
Abstract
The CCCTC-binding factor (CTCF), which anchors DNA loops that organize the genome into structural domains, has a central role in gene control by facilitating or constraining interactions between genes and their regulatory elements. In cancer cells, the disruption of CTCF binding at specific loci by somatic mutation or DNA hypermethylation5 results in the loss of loop anchors and consequent activation of oncogenes. By contrast, the germ-cell-specific paralogue of CTCF, BORIS (brother of the regulator of imprinted sites, also known as CTCFL)6, is overexpressed in several cancers, but its contributions to the malignant phenotype remain unclear. Here we show that aberrant upregulation of BORIS promotes chromatin interactions in ALK-mutated, MYCN-amplified neuroblastoma10 cells that develop resistance to ALK inhibition. These cells are reprogrammed to a distinct phenotypic state during the acquisition of resistance, a process defined by the initial loss of MYCN expression followed by subsequent overexpression of BORIS and a concomitant switch in cellular dependence from MYCN to BORIS. The resultant BORIS-regulated alterations in chromatin looping lead to the formation of super-enhancers that drive the ectopic expression of a subset of proneural transcription factors that ultimately define the resistance phenotype. These results identify a previously unrecognized role of BORIS—to promote regulatory chromatin interactions that support specific cancer phenotypes..
Contribution: Design, processing, analyzing and interpretation of scRNAseq, micro-array, ChIP-seq, HiChIP and public data. Discussing, writing and editing of paper.