Giotto, a pipeline for integrative analysis and visualization of single-cell spatial transcriptomic data.
Abstract
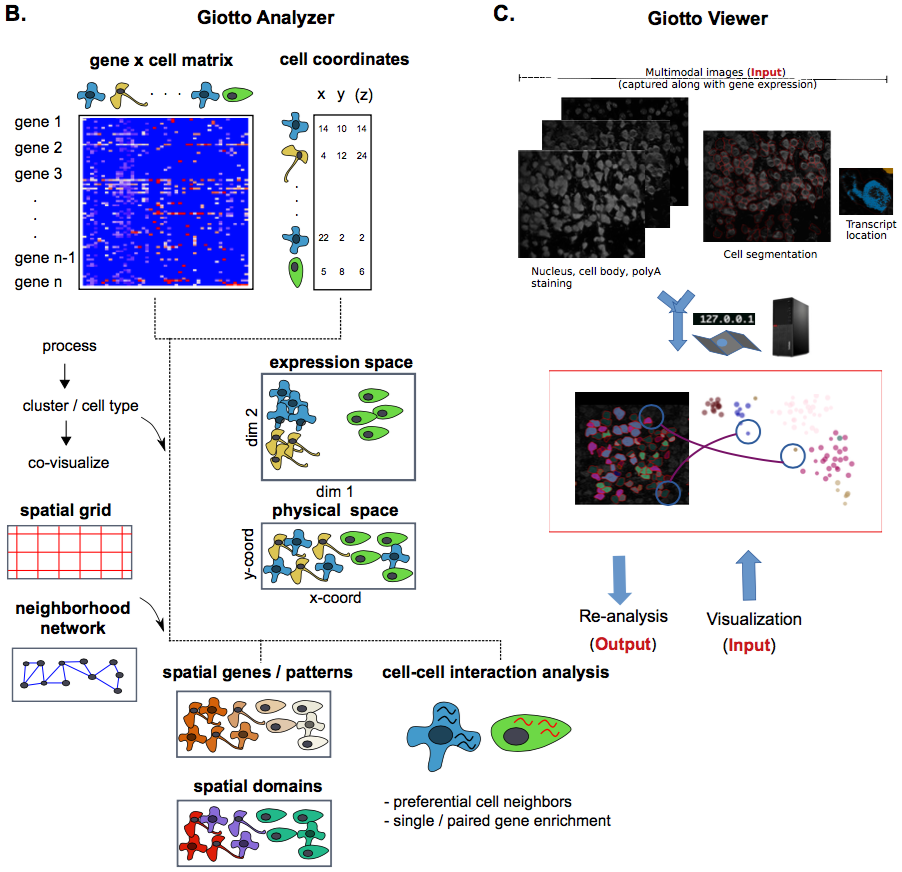
The rapid development of novel spatial transcriptomics technologies has provided new opportunities to investigate the interactions between cells and their native microenvironment. However, effective use of such technologies requires the development of innovative computational algorithms and pipelines. Here we present Giotto, a comprehensive, flexible, robust, and open-source pipeline for spatial transcriptomic data analysis and visualization. The data analysis module implements a wide range of algorithms ranging from basic tasks such as data pre-processing to innovative approaches for cell-cell interaction characterization. The data visualization module provides a user-friendly workspace that allows users to interactively visualize, explore and compare multiple layers of information. These two modules can be used iteratively for refined analysis and hypothesis development. We illustrate the functionalities of Giotto by using the recently published seqFISH+ dataset for mouse brain. Our analysis highlights the utility of Giotto for characterizing tissue spatial organization as well as for the interactive exploration of multi- layer information in spatial transcriptomic and imaging data. We find that single-cell resolution spatial information is essential for the investigation of ligand-receptor mediated cell-cell interactions. Giotto is generally applicable and can be easily integrated with external software packages for multi-omic data integration.
Contribution: Design and build of Giotto Analyzer. Discussing, writing and editing of paper.